Methods
All-atom molecular dynamics simulation, molecular docking, and various other theoretical and computational techniques for the analysis of complex systems.
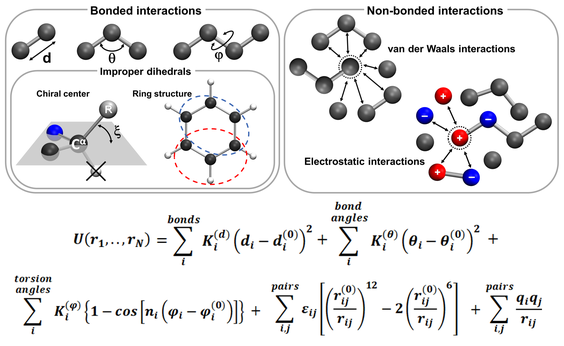
Molecular dynamics
Molecular dynamics (MD) is a technique used to simulate the motion of a complex molecular system at the atomic scale. Particles are governed by relatively simple interaction potentials, and the time evolution of their positions follows the classical equations of Newton. Thus, MD simulation is a sampling methodology that obeys the basic principles of statistical mechanics.
This technique can be applied to virtually any type of molecular system. In particular, it is often used to discover hidden details in the working mechanisms of biological molecules. In the case of proteins, MD simulation can be used to study the dynamic properties of their structures, either alone or in interaction with any type of molecular partner.
Molecular docking
Molecular docking predicts the binding location and affinity of a guest molecule interacting with a target host. The prediction is obtained by combining a search algorithm, which samples different conformations of the guest bound on the host surface, and a scoring function, which provides an empirical estimate of the binding energy in each conformation.
Although being a 'static' technique, with only a limited possibility of accounting for the rearrangement of the host in the binding process, molecular docking can often capture the essential features of the structure of a molecular complex. In the practice, this methodology is frequently used to predict the association of small ligands to target macromolecules.
Bioinformatic analysis tools
Biological data encoded in molecular systems are often hidden in their complex structure and dynamics. Many types of software tools have been developed to extract and interpret these data, by implementing algorithms that rely on solid physical and chemical principles, and on mathematical and statistical methodologies for their analysis.
A typical example is the amino acid sequence of a protein, which possesses key molecular features (e.g., size, charge/polarity, hydrophobicity/hydrophilicity, etc.) that ultimately determine its structure. By combining this wealth of information, further properties can be predicted, such as the order/disorder propensity or the binding affinity towards potential molecular partners.